Research Projects
Evolutionary simulations of somatic mosaicism
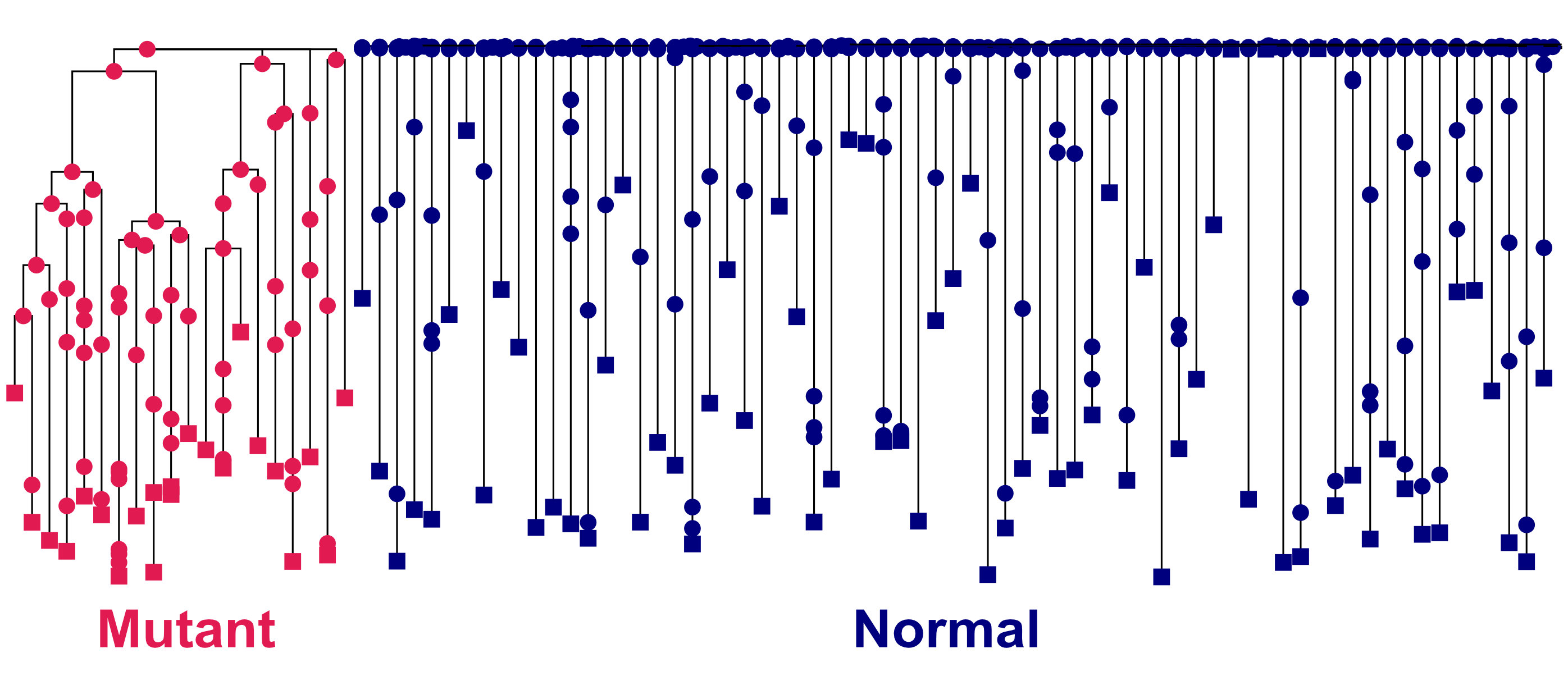
The cells in our bodies accumulate somatic mutations throughout our lives. Mutations with fitness benefits can cause a cell to overproliferate, creating a clonal lineage of daughter cells — a phenomenon that can eventually lead to cancer.
I collaborated with the Armanios lab at the JHU School of Medicine to develop an agent-based model of somatic evolutionary dynamics in the blood, built with the population genetics software SLiM.
My simulation tracks the development of clonal lineages in the blood over time, accounting for how telomere shortening can induce cell death. Our model showed that telomere length alone can affect an individual's risk of accumulating harmful somatic mutations and cancer.
Publications
DeBoy EA*, Tassia MG*, Schratz KE, Yan SM, et al. (2023). Familial clonal hematopoiesis in a long telomere syndrome. New England Journal of Medicine, 388: 2422–2433.
Telomere-to-Telomere Consortium
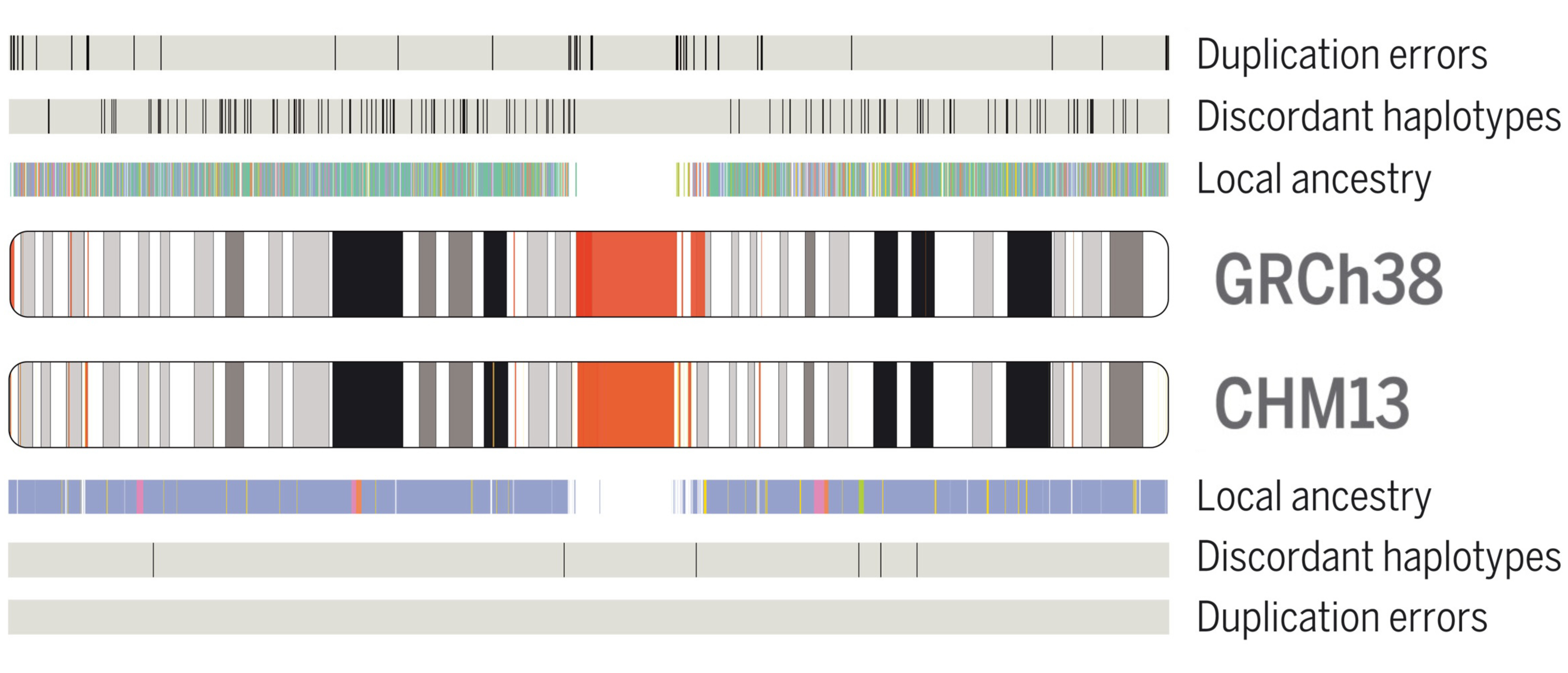
When the human genome was first sequenced in 2001, 8% of its most complex and repetitive regions were inaccessible to current sequencing technologies, and remained incomplete for the next two decades.
In 2021, the Telomere-to-Telomere Consortium used long-read sequencing to assemble the first ever complete human reference genome, T2T-CHM13.
Within this collaborative project, I led the analysis of novel variants in the T2T-CHM13 assembly to identify population-specific genetic variation. These discoveries highlight the potential impact of the T2T-CHM13 reference for revealing novel insights into human evolutionary history. I also annotated the local ancestry and Neanderthal-introgressed regions of the genome to facillitate its use as a resource for the scientific community.
Publications
Soto DC*, Kirsche M*, Yan SM*, & Zarate S*. (2023). The human reference genome is finally complete. The Science Breaker.
Aganezov S*, Yan SM*, Soto DC*, Kirsche M*, Zarate S*, et al. (2021). A complete reference genome improves analysis of human genetic variation. Science, 376(6588): 54.
Nurk S*, Koren S*, Rhie A*, Rautiainen M*, ... , Yan SM, et al. (2022). The complete sequence of a human genome. Science, 376(6588): 44–53.
Local adaptation at human structural variant loci
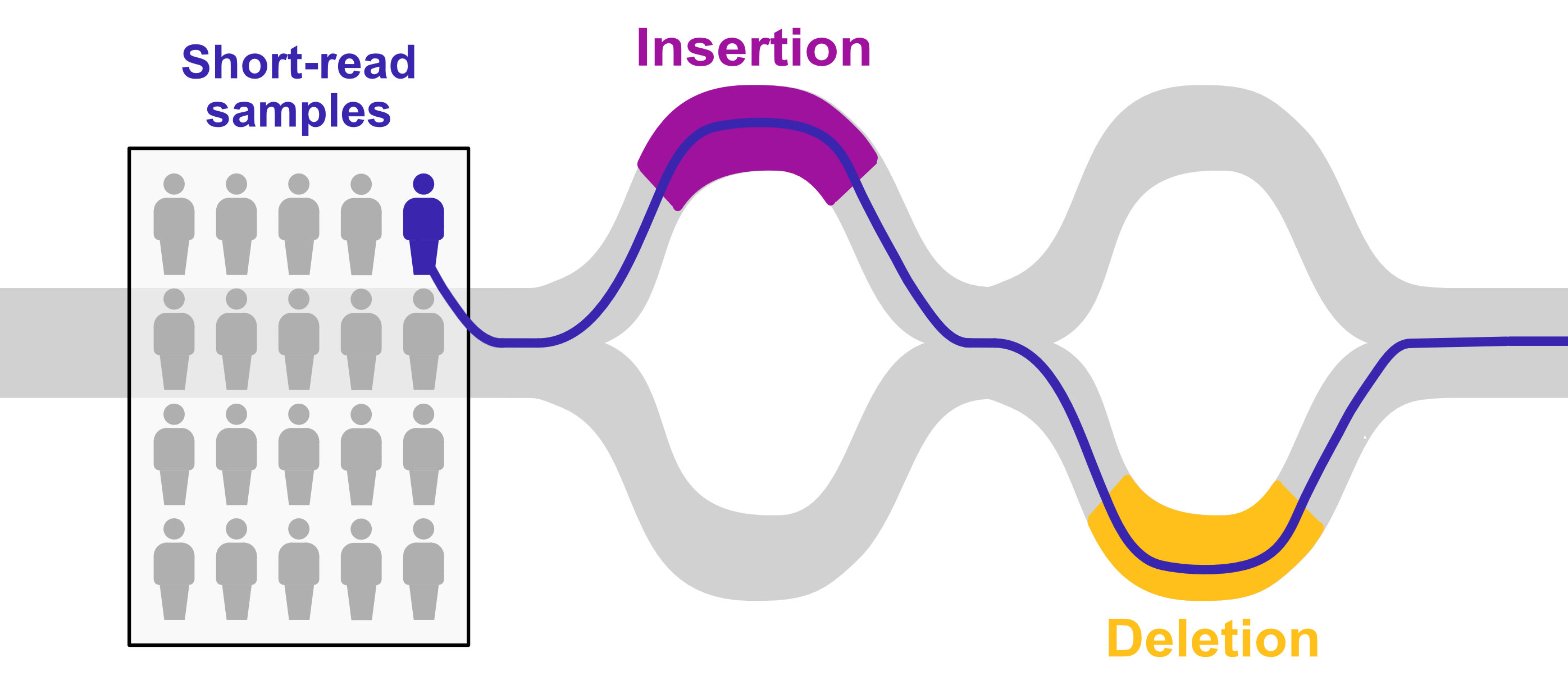
Large genomic rearrangements (insertions, deletions, etc.), called structural variants (SVs), are an understudied form of genetic variation that can have dramatic functional impacts.
These variants are hard to discover with normal sequencing methods, and instead require expensive and low-throughput long-read sequencing to study comprehensively. We used a hybrid approach to study structural variants on a population-wide scale: discovery with long-read sequencing, followed by graph genotyping in a large dataset of diverse human individuals.
We found evidence of 220 structural variants under population-specific positive selection, including a sequence in the immunoglobulin locus that was inherited into humans from Neanderthals.
Publications
DeGorter MK*, Goddard PC*, Karakoc E, Kundu E, Kundu S, Yan SM, et al. (2023). Transcriptomics and chromatin accessibility in multiple African population samples. bioRxiv.
Yan SM, Sherman RM, Taylor DJ, Nair DR, Bortvin AN, Schatz MC, & McCoy RC. (2021). Local adaptation and archaic introgression shape global diversity at human structural variant loci. eLife, 10: e67615.
Archaic hominin gene expression
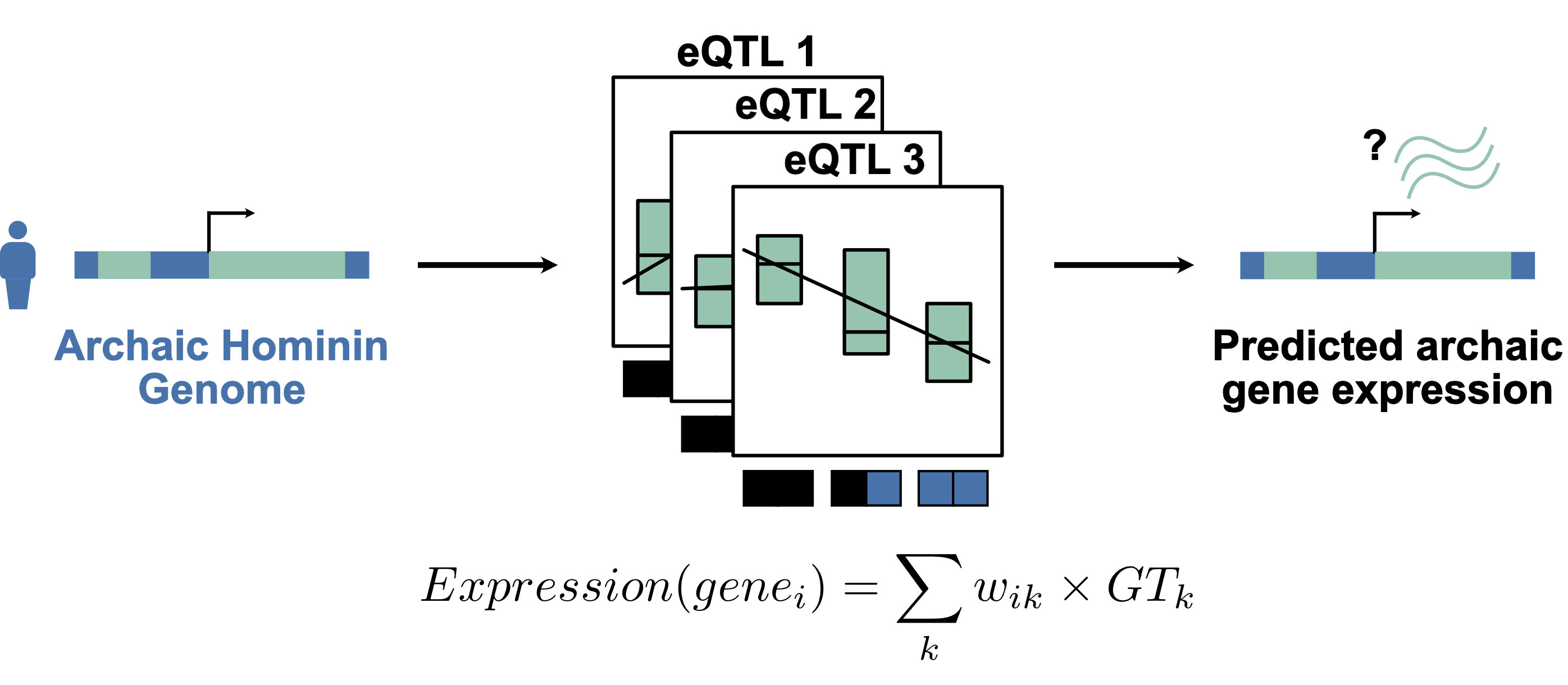
Recent research on Neanderthals and Denisovans, the closest evolutionary relatives to modern humans, has begun exploring patterns of gene expression to understand how these archaic hominins differed from modern humans.
Studying gene expression means working on RNA, which is too fragile to survive the tens of thousands of years that separate us from archaic hominins. Consequently, researchers have developed creative approaches for inferring archaic gene expression from DNA sequencing data, which I described in this review.
Publications
Yan SM & McCoy RC. (2020). Archaic hominin genomics provides a window into gene expression evolution. Current Opinion in Genetics & Development, 62: 44-49.
Yan SM & McCoy RC. (2019). Functional divergence among hominins. Nature Ecology & Evolution: News & Views, 3: 1507–1508.